Computational studies of Ni(II) photosensitizers complexes containing 1,1′-bis(diphenylphosphino)ferrocene and dithio ligands
Résumé
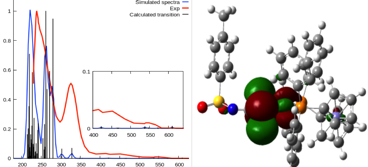
Detailed theoretical studies of Ni(II) complexes in a distorted square planar form and containing dithio and (P,P) chelating ligands were performed. These Ni(II) complexes are investigated for their use in dye-sensitized solar cells (DSSC). Structures and UV-Vis spectra are calculated at density functional theory (DFT) and time-dependent density functional theory (TD-DFT) theories using B3LYP and CAM-B3LYP functionals and 6-31G(d,p) and 6-31G+(d) basis set. Geometry optimizations result in excellent agreement with the experimental results. Moreover, the analysis of the frontier molecular orbitals (FMOs) allowed a detailed assignment and a clear analysis of the electronic transitions. The TD-DFT calculations reproduce the main spectroscopic properties observed and substituent effects. The results reveal that all absorption spectra are characterized by mixed character mainly dominated by metal to ligand and ligand to ligand charge transfer (MLCT and LLCT). We unveil how the substituent variations affect the DSSCs features of the complexes.
Nous avons mené des études théoriques détaillées sur des complexes de Ni(II) de structure plan carré déformé portant des ligands chélatants dithio et bis-phosphino. Ces études nous ont permis d’évaluer la possibilité d’utiliser ces complexes de Ni(II) dans des cellules solaires à pigment photosensible (DSSC). Nous avons modélisé la structure et calculé les spectres UV–vis de ces complexes selon la théorie de la fonctionnelle de la densité (DFT) et la DFT dépendante du temps (TD-DFT) en utilisant les fonctionnelles B3LYP et CAM-B3LYP avec les bases 6-31G(d,p) et 6-31G+(d). Les résultats des optimisations géométriques montrent une excellente concordance avec les résultats expérimentaux. De plus, l’analyse des orbitales moléculaires frontières (OMF) a permis de comprendre clairement et d’assigner en détail les transitions électroniques. Les calculs TD-DFT reproduisent les principales propriétés spectroscopiques observées et les effets des substituants. Selon nos résultats, tous les spectres d’absorption de ces complexes révèlent un caractère mixte dominé principalement par les transferts de charge métal vers ligand et ligand vers ligand. Nous présentons la façon dont les variations des substituants influent sur les propriétés des cellules solaires à pigment photosensible composées de ces complexes
Fichier principal
 bs-second-revised-paper-cjc-03-02-20SH-ar.pdf (1.64 Mo)
Télécharger le fichier
hal-02512291-graphabstr.png (42.38 Ko)
Télécharger le fichier
bs-second-revised-paper-cjc-03-02-20SH-ar.pdf (1.64 Mo)
Télécharger le fichier
hal-02512291-graphabstr.png (42.38 Ko)
Télécharger le fichier
{kind=link}
Origine : Fichiers produits par l'(les) auteur(s)
Loading...